



pdf Stormy Chamberlain, PhD Popular
By Administrator 412 downloads

In conversation with Stormy Chamberlain, PhD On our virtual tour through the Angelman community, we stopped at the University of Connecticut to meet Stormy Chamberlain, PhD. In 2009 she established her own lap at Uconn, focused on human imprinting disorders, such as Angelman syndrome. With good reasons she is member of ASF Scientific Advisory Committee, because time and again she surprises the community with groundbreaking insights. Especially her research dealing with pluripotent stem cells usher in a new era of Angelman drug development. High time for us to inquire more precisely …
When did you first come into contact with Angelman syndrome? What makes AS so special from a scientific point of view?
I first became aware of Angelman syndrome as a PhD student at the University of Florida in 1997. My thesis project involved finding the genes important for Prader- Willi syndrome using a mouse model, and I had to learn about and understand the reciprocal regulation of paternal and maternal copies of chromosome 15q.
What makes Angelman syndrome interesting for you from a scientific point of view?
I have always been intrigued as to why evolution chose to turn off one of the copies of a gene that is so important for normal development. All placental mammals imprint UBE3A and it’s not clear why. One might argue that the dose of the gene is super important, but actually, expression from the maternal copy of UBE3A is increased over the course of neurodevelopment, just as the paternal copy is getting silenced, so the dosage argument doesn’t make a lot of sense. Although every placental mammal imprints UBE3A, there are subtle differences in how it achieved as well. This means that the imprinting is important, but evolution has come up with some slightly different ways to achieve it. I just think that the complexity of how this all happens is so interesting, and can reveal lessons that can be applied to other genes in the genome.
„I am committed to AS research because I firmly believe that by investing in knowledge about a disorder and gene that science can make a therapeutic that is more likely to provide substantial benefit. AS will be an excellent example for other disorders.“
One part of your research is focused on pluripotent stem cells. What exactly is it?
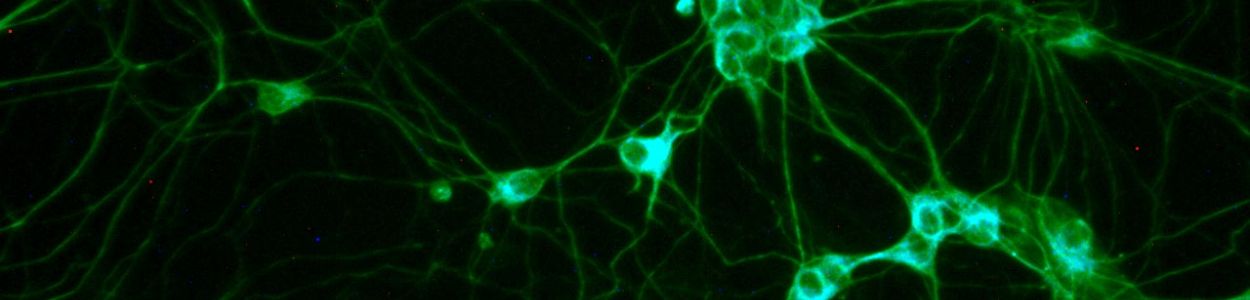
My lab uses human pluripotent stem cells as a model system. Some people use mice to study Angelman syndrome, but my lab uses human pluripotent stem cells. These cells can be converted into neurons, so we can study actual human neurons with the disorder. Furthermore, we can follow the process of imprinting during neurodevelopment. The stem cells express UBE3A from both parental copies, but as they are converted into neurons, the paternal copy of UBE3A becomes silenced. We study this process. The other cool feature about these cells is that if we use induced pluripotent stem cells (as opposed to human embryonic stem cells)—where we convert a patient blood cell or a skin cell into a pluripotent stem cell--they carry the genetics of the patient.
Which role does pluripotent stem cells play for research on Angelman syndrome?
We can use pluripotent stem cells to study the process of imprinting during human neurodevelopment. This helps us understand how the different parts of the human genome regulate this process. We can also use the neurons created from the pluripotent stem cells to understand the deficits in early human neurons with Angelman syndrome. This is important to figure out what goes wrong first, so we can determine how loss of UBE3A causes it. It’s important to do this in human cells, since this also might differ between human and mouse. Finally, we have been using our pluripotent stem cell models to screen therapeutics that can activate paternal UBE3A. We have tested small molecules (such as topotecan), antisense oligonucleotides (ASOs), and shRNAs. The therapeutics that are DNA-based drugs, like ASOs and shRNAs, need to be tested in a human cell model because the DNA is different between humans and mice. These pluripotent stem cells are pretty much required for testing these therapeutics.
How are pluripotent stem cells produced?
Many of our pluripotent stem cells are produced using reprogramming. We take a blood sample or skin sample from a patient and artificially express stem cell genes (OCT4, KLF4, SOX2, and MYC) using a viral vector. These genes jumpstart the stem cell program in the blood or skin cells, turning them into stem cells. We can also make Angelman syndrome-like deletions—either the large deletions seen in patients or smaller, specific mutations of UBE3A—using CRISPR/Cas9 to cut the DNA in human embryonic stem cells. This means that we can create identical twin stem cells with and without Angelman syndrome.
How do you assess the possibility of using pluripotent stem cells as a possible therapeutic approach for the treatment of Angelman syndrome?
There is no possibility of using the stem cells as a therapeutic approach for AS. The brains of individuals with AS develop with normal structure and cell types overall. If we introduce new stem cells or neural stem cells, they are not likely to make the connections that they’re supposed to in order to be of benefit to the patient. Furthermore, the dangers of putting new cells into a brain is much greater than the anticipated benefit. There are much better and safer therapeutic approaches for AS.
„My mission is to use my talents and tools to learn all we can about AS and contribute to a variety of therapeutic approaches. Along the way, my mission is to train the next generation of researchers who will do the same for AS and other disorders. “
Neuron models can be made of pluripotent stem cells. How can these neuron models help to understand Angelman syndrome? Could neuron models be developed for all AS genotypes? What added value could this bring to the AS research?
The neurons made from pluripotent stem cells can help us understand human gene regulation—how UBE3A and UBE3A-ATS are regulated during neural development. They can help us understand what are the earliest deficits in the neuron. If we can figure out how UBE3A causes the first deficits, those are the most critical to fix because other things are likely downstream of the primary deficits. They can also be used to test therapeutics. These pluripotent stem cell models can be made for all AS genotypes. We have already made them for deletions, multiple UBE3A mutations, and uniparental disomy. The only one we don’t have is imprinting defect. It’s valuable to have the different genotypes for a variety of reasons. First, the deletion type has really helped us look at regulation because there is only the paternal copy present. This has helped us understand how UBE3A-ATS silences UBE3A. The UBE3A mutation type really helps us narrow down which neuron deficits are caused by UBE3A (as opposed to loss of one copy of the other genes). One of our cell lines with a UBE3A mutation has revealed a new way to regulate UBE3A protein as well. The uniparental disomy type helps us understand whether those individuals make a little bit of UBE3A, which might give them different therapeutic options. We have also used uniparental disomy neurons to see whether therapeutic approaches turn on both paternal copies of UBE3A, which can make too much UBE3A a possibility. The cells have indicated that they may want to use a lower dose of ASO on individuals with uniparental disomy and imprinting defect (which also has 2 copies of UBE3A that act like they’re paternal).
What role could neuron models play in the success of clinical trials? (e.B. ASOs, AVV gene therapy, Neuren, OV101)
Our models aren’t as helpful for OV101 and Neuren approaches because it’s hard to extrapolate from a neuron in a dish to an organism. This might change as we get better at making organoids or mini-brains, which may have features more like actual brains. However for ASOs they can help and have already helped us identify the sequence to target and learn the “rules” for the drug. For instance, how much UBE3A can the paternal allele maximally produce? How do the drugs work? What other genes do they impact? For AAV gene therapy, they can help us anticipate how much UBE3A is produced in a cell. They’ve also told us a bit about how much of each UBE3A protein isoform is normally in a human neuron and where in the cell it needs to be.
You also do research on shRNA together with Ovid pharmaceuticals. Can you explain briefly how shRNA maybe one day could be used as treatment for AS?
The shRNA approach works a lot like ASOs—they cut UBE3A-ATS RNA at a specific spot. However, they can be delivered with AAV vectors, so it’s a long-lasting approach that can’t potentially over-produce UBE3A, like other types of AAV gene therapy can. It’s also a proof-of-concept for other RNA cutters that can be similarly delivered, such as miRNA, CRISPR/Cas13, and ribozymes. They all cut RNA, and can theoretically be delivered using long lasting viral vectors. Our goal for the shRNA is to use it to provide a one-time treatment that would last decades. We’ll have to be extra careful to think about safety, though because it is so long lasting.
How do you assess - from today's point of view - the future of the drug treatment of Angelman syndrome?
Like you all, I’m anxiously awaiting the results of the clinical trials for ASOs. I think we need to be patient and wait until kids of different ages have been treated with what is determined to be the best dose before we get too confident. Other therapeutic approaches, like the shRNAs, will depend on how the ASO trials go. However, I think we all should be optimistic because we have three different attempts coming up and their interest and progress has drawn other companies into Angelman therapeutics. Pharma thinks they can make a difference for kids with AS, and it’s always good to have their optimism behind the community. If the ASOs work, then there will be many trying to make better therapeutics. Better can mean many things—easier to deliver, longer lasting, or targeting a different area of the brain. I just hope that some companies and research labs continue to study ways to therapeutically help older individuals with AS who may not benefit as strongly from the ASOs and other genetic therapies.
How do you assess the developmental potential of "adult Angelman brains" in a possible future treatment?
The information inferred about this from mice is only a hint, and shouldn’t be taken too literally. Humans take much longer to develop, so it seems possible that humans would have a longer developmental window for treatment. The only way to know this for sure is to treat individuals of different ages with ASOs. I suspect that will happen by real, careful clinical trials, however, it will likely happen after trials involving younger kids have been completed. Humans can learn throughout life, so it’s possible that there will be some benefit of restoring UBE3A in older individuals, too. Then it becomes a risk versus benefit calculation.
„For the future of Angelman patients, I hope we change what it means to have a diagnosis of Angelman syndrome. I hope we can provide therapeutic options to make life easier for a child with AS and their families.“